Test drivin' rDock
rDock is an ope n source docking program, trying to solve the same kind of scientific questions as Autodock Vina, which I have covered in a couple of earlier blogposts. It was open sourced in 2012, but development of the earliest versions dates back to 1998. More details can be found on the rDock homepage.
n source docking program, trying to solve the same kind of scientific questions as Autodock Vina, which I have covered in a couple of earlier blogposts. It was open sourced in 2012, but development of the earliest versions dates back to 1998. More details can be found on the rDock homepage.
installation was done via SVN checkout and compiling. For me it was straightforward on my Linux workstation:
#Prerequisites sudo apt-get install libcppunit-1.13-0 libcppunit-dev libpopt0 libpopt-dev python-openbabel #Get code and compile svn checkout svn://svn.code.sf.net/p/rdock/code-0/trunk rdock-code-0 cd rdock-code-0/build make linux-g++-64 make test #Clean up temporary build files after succesful test make clean
Before usage a few environment variables needed to be set, so the following lines were added to .bashrc:
export RBT_ROOT=/path/to/rDock/installation/ export LD_LIBRARY_PATH=$LD_LIBRARY_PATH:$RBT_ROOT/lib export PATH=$PATH:$RBT_ROOT/bin
For other architectures and details about the installation check out the guide: http://rdock.sourceforge.net/installation/
For the test I chose the same receptor-ligand complex as I have previously been testing Autodock Vina with: 1OYT. This time the docking program needs a SD file for the ligand as input and a .mol2 formatted file with assigned Sybyl atom types. The authors provide a test-set with various correctly formatted files, but I chose to take the original source from the Astex diverse set.
the protein.mol2 and the ligand.mol was copied to a seperate directory and renamed for further processing. The format of the ligand was converted with obabel:
obabel ligand.mol -O 1oyt_ligand.sdf
The rDock program need a parameter file with setting for the generation of the cavity definition and scoring function for the docking, so the following was added to the plain text file 1OYT_redock.prm
RBT_PARAMETER_FILE_V1.00 TITLE 1OYT_redock_test RECEPTOR_FILE 1oyt_astex.mol2 RECEPTOR_FLEX 3.0 ################################################################## ### CAVITY DEFINITION: REFERENCE LIGAND METHOD ################################################################## SECTION MAPPER SITE_MAPPER RbtLigandSiteMapper REF_MOL 1oyt_ligand.sdf RADIUS 6.0 SMALL_SPHERE 1.0 MIN_VOLUME 100 MAX_CAVITIES 1 VOL_INCR 0.0 GRIDSTEP 0.5 END_SECTION ################################# #CAVITY RESTRAINT PENALTY ################################# SECTION CAVITY SCORING_FUNCTION RbtCavityGridSF WEIGHT 1.0 END_SECTION
This is standard settings for the docking program, using a reference ligand to define the cavity. The only change needed was the insertion of the receptor file name and the reference ligand name.
rbcavity -r 1OYT_redock.prm -was > 1OYT_redock_cavity.log
To parse the mol2 file and generate the necessary cavity definitions and grid files, the utility rbcavity is used. The output specified with -was is a 1OYT_redock.as file which is needed for the next step.
rbdock -r 1OYT_redock.prm -p dock.prm -n 100 -i 1oyt_ligand.sdf -o 1oyt_redock_out > 1oyt_redock_out.log
We reuse the docking setup file 1OYT_redock.prm and additionally specify a docking script “dock.prm”. This file is a standard file found in the
$RBT_ROOT/data/scripts directory. It contains information about how to stage the docking with different weights at different times during the docking. The docking run is single threaded. This is a bit surprising in these parallel times and as the genetic algorithms underlying the actual docking are somewhat simple to parallelize. This may be an issue with technical debt, as the docking programs first versions date back to 1998. Well before multi threaded processors became common at desktop computers. the -n 100 switch sets that 100 docking runs should be made. On my workstation this takes a couple of minuttes. The output is an SD formatted file with the Score and other information set as properties. Luckily the rDock utility sdsort can sort the output file after a specified property
sdsort -n -f'SCORE' 1oyt_redock_out.sd > 1oyt_redock_out_sorted.sd
The -n set the sorting to be numerically. As I’m interested in the docking accuracy, the sdrmsd utility comes in handy. It compares the poses in the sd output file with the specified reference, taking into account equivalent atoms in rotatable groups like e.g. Phenyl.
sdrmsd 1oyt_ligand.sdf 1oyt_redock_out_sorted.sd
POSE RMSD_NOFIT 1 0.54 2 0.80 3 0.74 4 0.47 5 0.45 ... 21 5.38 22 2.79
The 21 highest scoring poses, has a very low RMSD in comparison with the crystallographic reference. They can be inspected in Pymol
pymol 1oyt_astex.mol2 1oyt_redock_out_sorted.sd 1oyt_ligand.sdf
The video is played “backwards”, so the movie goes from lowest scoring to highest scoring.
with the sdrmsd utility its possible to write a new SD file with the RMSD field added, and the use the sdreport utility to generate a report with the properties of interest (here Score and RMSD).
sdrmsd 1oyt_ligand.sdf 1oyt_redock_out_sorted.sd -o 1oyt_redock_out_rmsd.sd sdreport -cSCORE,RMSD 1oyt_redock_out_rmsd.sd > scores.csv
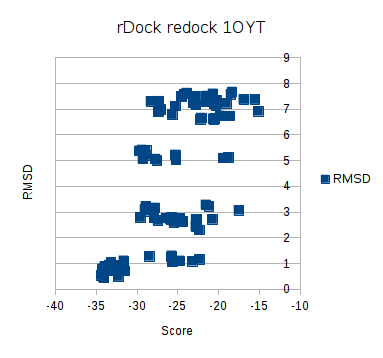
Plotting the RMSD as a function of the score, shows a nice grouping of the correct ones, with a clear seperation in score values to the wrong ones. A few of the poses < 1 Å have a medium score, even though they are correct according to the RMSD < 2Å criterion widely used to evaluate docking performance. However as demonstrated with the Vina series of blogpost, using redocking as a measure of docking accuracy can lead to overestimation of the docking power of the program. Next time I’ll see what happens with a cross docking experiment.
rDock in summary so far:
- PROS:
- Open Source
- Relatively Fast
- Really useful SD file handling utilities
- CONS:
- Setup files is more cumbersome in scripting.
- VERY sensitive to mol2 file format of receptor. e.g. Open Babel, Pymol and Chempy (via Pymol) generates mol2 files which results in warning and docking errors.
Pingback: Cross Docking with rDock | Wildcard Pharmaceutical Consulting