Learn how to hack RDKit to handle peptides with pseudo atoms
The chemoinformatics package Rdkit has is strength with handling small organic molecules. These molecules are characterized by a large diversity in chemical structures. A description of the exact way the atoms bond together are necessary to understand what molecule it is.
Biological macro molecules are often build from repeating sequences of standard building blocks, such as amino-acids or nucleotides. This makes it possible to specify the structure of a protein by writing its sequence of amino-acids, which has been coded into single letters. As example, the peptide met-enkefalin (https://en.wikipedia.org/wiki/Enkephalin) has the sequence Tyr-Gly-Gly-Phe-Met or YGGFM in single letter notation.
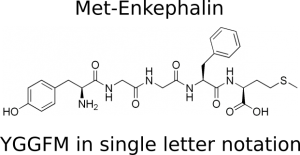
Met-Enkephalin as full structure and amino acid sequence
But what if there’s suddenly a need to work with modified or unnatural amino acids? Of course the whole structure can be written out like in the illustration, but as the sequence length increases this becomes too complex to overview. So instead the two worlds can be combined by defining pseudo atoms which represent the naturally occurring amino acids. The old drawing program ISIS/Draw has support for these kind of structures, as well as its possible to use in MarvinSketch. My good friend Jan’s proteax software also has support for writing peptides in condensed form with pseudo atoms, as well as functions that can inter convert between fully atomistic representations, mixed representations with pseudo-atoms and line-notation.
To cut a long story short, can Rdkit read Sdfiles with amino acids specified as pseudo atoms? No, not straight away. But with just a small modification it can.
But due to the open source nature, its relatively straightforward to extend the capabilities of Rdkit. ISIS/Draw defined the pseudo atoms as atoms with atomic numbers starting from 171. Its good the nuclear physicists are not that far yet 😉 Rdkit has its atom definitions in the file Code/GraphMol/atomic_data.cpp So the pseudo atoms need to be defined there. Below is a excerpt of the additions made to that file. First Column is the atomic number. Rdkit expects a continuous sequence, so dummy values have to be filled in for atom number 112 to 170. The follows the label, the covalent radius, the rB0 (Some born radius, the origin is lost in rdkit lore) , the van der Waals radius, the atomic mass, the number of outer shell electrons, the most common isotope and the most common isotope mass followed by the number of allowed valences. I put in the most sensible values i could think off and find from course grained force fields, and calculated the atomic mass and exact atomic mass with Rdkit for each amino acid (minus H2O). Cysteine is treated different, it doesn’t contain the sulfur atom and has 5 outer shell electrons and a valence of 3. That way the sulfur atom can be added as an atom and used for creating cysteine bridges in the condensed format.
171 Ala 1.9 2 5.0 71.079 6 300 71.03711378 2 \n" "172 Arg 1.9 2 6.6 156.189 6 300 156.101111004 2 \n \ 173 Asn 1.9 2 5.7 114.104 6 300 114.042927432 2 \n \ 174 Asp 1.9 2 5.6 115.088 6 300 115.02694302 2 \n \ 175 Cys 1.9 2 5.5 70.071 5 300 70.029288748 3 \n \ 176 Gln 1.9 2 6.0 128.131 6 300 128.058577496 2 \n \ 177 Glu 1.9 2 5.9 129.115 6 300 129.042593084 2 \n \ 178 Gly 1.9 2 4.5 57.052 6 300 57.021463716 2 \n \ 179 His 1.9 2 6.1 137.142 6 300 137,058911844 2 \n \ 180 Ile 1.9 2 6.2 113.16 6 300 113.084063972 2 \n \ 181 Leu 1.9 2 6.2 113.16 6 300 113.084063972 2 \n \ 182 Lys 1.9 2 6.4 128.175 6 300 128.094963004 2 \n \ 183 Met 1.9 2 6.2 131.2 6 300 131.040484908 2 \n \ 184 Phe 1.9 2 6.4 147.177 6 300 147.068413908 2 \n \ 185 Pro 1.9 2 5.6 97.117 6 300 97.052763844 2 \n \ 186 Ser 1.9 2 5.2 87.078 6 300 87.0320284 2 \n \ 187 Thr 1.9 2 5.6 101.105 6 300 101.047678464 2 \n \ 188 Trp 1.9 2 6.8 186.214 6 300 186.07931294 2 \n \ 189 Tyr 1.9 2 6.5 163.176 6 300 163.063328528 2 \n \ 190 Val 1.9 2 5.9 99.133 6 300 99,068413908 2 \n \ 191 Sec 1.9 2 5.5 70.071 5 300 70.029288748 3 \n \ 192 Pyl 1.9 2 6.4 237.303 6 300 237.147726848 2 \n \ 193 Xaa 1.9 2 4.5 57.052 6 300 57.021463716 2 \n "; 188 Trp 1.9 2 6.8 186.214 6 300 186.07931294 2 \n \ 189 Tyr 1.9 2 6.5 163.176 6 300 163.063328528 2 \n \ 190 Val 1.9 2 5.9 99.133 6 300 99,068413908 2 \n \ 191 Sec 1.9 2 5.5 70.071 5 300 70.029288748 3 \n \ 192 Pyl 1.9 2 6.4 237.303 6 300 237.147726848 2 \n \ 193 Xaa 1.9 2 4.5 57.052 6 300 57.021463716 2 \n ";
A fully patched rdkit is available from https://github.com/DrBjerrum/rdkit
After compiling the new version of RDKit it is possible to import Sdfiles with Pseudo Atoms.
condensedmol = Chem.MolFromMolFile('condensed_molfile.mol')
Draw.MolToFile(condensedmol, 'condensed_structure.png', size=(600,400), kekulize=True, wedgeBonds=True, imageType=None, fitImage=True)
Gives a nice litte picture of a molecule in mixed representation.
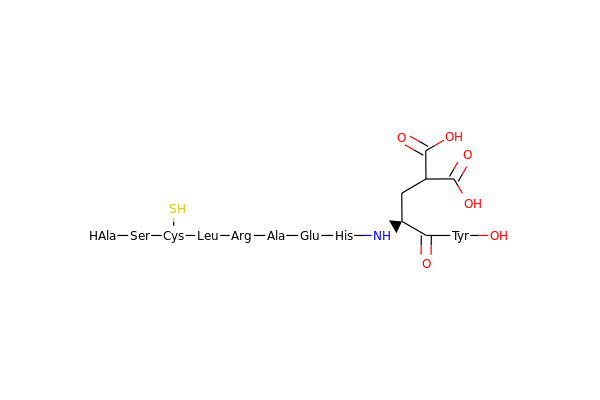
Lets try a slightly larger example. Insulin.
from rdkit import Chem
from rdkit.Chem import Draw
mol = Chem.MolFromMolFile('condensed_insulin.mol')
#Draw.ShowMol(mol,size=(600,600))
Draw.MolToFile(mol,
'condensed_insulin.png', size=(800,800), kekulize=True,
wedgeBonds=True, imageType=None, fitImage=True)
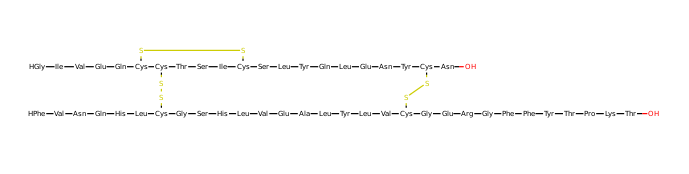
The drawing is a bit fuzzy, but in principle it works.
Smiles doesn’t work. It only parses two letters for atoms.
In [27]: mol2 = Chem.MolFromSmiles('[AlaH2]')
[13:29:20] SMILES Parse Error: syntax error for input: '[Ala]'
However SMARTS does if the “atomic number” of the pseudo atom is used.
In [28]: mol2= Chem.MolFromSmarts('[#171]')
In [29]: Chem.MolToSmiles(mol2)
Out[29]: '[AlaH2]'
By using the SMARTS mol object, substructure searching is possible.
In [34]: mol.HasSubstructMatch(mol2) Out[34]: True
Calculation of molecular mass works
In [42]: from rdkit.Chem import Descriptors In [43]: Descriptors.MolWt(mol) Out[43]: 5807.670999999996
Close to the value 5807.57 i found online (http://www.scbt.com/datasheet-360248.html)
Writing to Molfile works
In [32]: Chem.MolToMolFile(mol, 'ala.mol')
RDKit
1 0 0 0 0 0 0 0 0 0999 V2000
0.0000 0.0000 0.0000 Ala 0 0 0 0 0 0 0 0 0 0 0 0
M END
Some of the fingerprints and descriptors seemingly work. But please do check that they do something sensible before using them for production or research work, I have not tested them. But this opens up a whole bunch of interesting possibilities. Alignment free comparison of protein similarity would be usable I think, Let me know if you have other ideas or investigate it further.
In [38]: from rdkit.Chem import rdMolDescriptors In [39]: rdMolDescriptors.CalcMolFormula(mol) Out[39]: 'H4AlaArgAsn3Cys6Gln3Glu4Gly4His2Ile2Leu6LysO2Phe3ProS6Ser3Thr3Tyr4Val4' In [47]: morgan = rdMolDescriptors.GetMorganFingerprintAsBitVect(mol,2) In [48]: morgan.ToBitString() Out[48]: '00000001000100000000000001000010000001100000000000000000000000001010000000000000000000000000000000000000000000010000000000001000011000000100000000000100000000101001000000000000000000000000000000000100000000000000000000000010000000000010000010100000000000000000000000000000000000000000000000001000000010000000100000000000100000000000001000000100000001000100000000100001000000000000010000100001000100000000000001000010000000001000000000000000000000000000000000000000000000000000010000000000000000000000000000000000100000000000000000000000000000000000000000100000000000000000000000000000000000000000000000000000000000100000000000000000000000000100000000000000000010000000000000010000000010100000000000000000000100000000000000100000000000000000000000000001000001010000000000000000000000000100100000000000000000010000000000000000000001000000000000000000010001001000000000000010000000000000000000000010000000000010000000000000110000000000000001000000000000000000000000000000010000000010000000000000010000000010000000000000000000000000000000000000000000100000000001000000000101000000000000000000000000000010000001000000000000000101000000001000000000000001001000000000100000000100000000000000000000100000100000000000010000000000000000000000010000000000000000000000000000000000000000000000000000100000000000001100001000000000000000000000010010000000000000000000000000000000001001000000000000000000000000000010000110000000000000000000000000010000000000000000000010000000001000000100000000000000000000000000000100000000000000000000000000000000100100000000010000000010100000000001000000000000000000010000000000000001000000000100100000000000000000000000000000000000000000000000000000000000000000000000000000000000100000000000000000000000000010000100100000000100000000000000000000000000000000000000000000000000001001000000001000000000000000000110000000000000000010000000000000001000000101000000000000000000000000010000000000000000000000000001000000000000000000001000000001000000000000000000000000000000001000000000010000000000000000000000001000000000000000000010'
So a lot of the usual RDkit goodies seem to work. To be truly useful, it should be possible to inter convert between full structure and condensed structure. On the other hand Proteax already has this capability, and the idea was originally to just ensure interoperability. Please comment below if you find it useful and how you used it.
Best Regards
Esben
P.S: Some examples of condensed molfiles to play with.
Examples_Condensed_Molfiles