Ligand docking with Smina
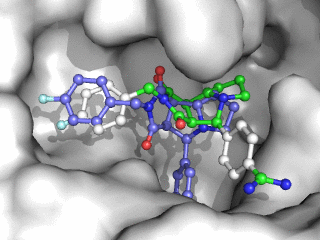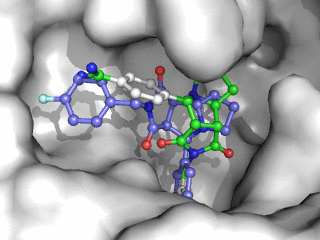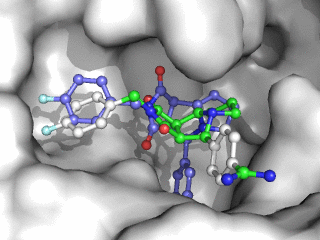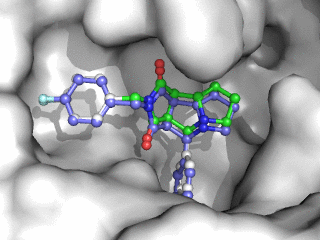
Moleculer Docking is a powerful technique for studying potential ligand-receptor interactions. It can be done with free tools. In this blog post I showcase the docking program Smina, a fork of Autodock Vina.
Automated docking is the process of prediction of how one molecule binds to another in a stable complex. For small drug-like molecules this process has been used in drug discovery project for evaluation of proposed ligands and also for in silico screening of virtual compound libraries to find potential ligands for a protein target with a known molecular structure.
In this blog post I’ll take a free program for a test drive. Autodock Vina (http://vina.scripps.edu/) is an open-source program for molecular docking of small ligands to protein targets. Since it was open sourced there have been a couple of forks. Quick Vina 2 aims at accelerating Autodock Vina , VinaLC added MPI for parallel execution on clusters, and Smina added better control of scoring terms as well as a range of convenience functions for easy use from the command line (https://sourceforge.net/projects/smina). Smina, open-babel and PyMOL will be used. Smina comes with a static version for Linux, so it should just be placed in the path of the command line, No dependicies and libraries 😉
For this test the PDB file 1OYT is used. It is a complex between RECOMBINANT HUMAN THROMBIN and FLUORINATED INHIBITOR. Thrombin is involved in the coagulation cascade and an inhibitor for this protein could potentially as a “blood thinner” to prevent or dissolve blood cloths in stroke and to prevent myocardial infarction.
Pymol is used to download the PDB file, remove water molecules and split it into the ligand and receptor molecule.
fetch 1OYT remove resn HOH h_add elem O or elem N select 1OYT-FSN, resn FSN #Create a selection called 1OYT-FSN from the ligand select 1OYT-receptor, 1OYT and not 1OYT-FSN #Select all that is not the ligand save 1OYT-FSN.pdb, 1OYT-FSN save 1OYT-receptor.pdb, 1OYT-receptor
Previously I have always used the prepare_receptor4.py and prepare_ligand4.py
scripts that come as part of Autodock Tools, but newer versions of numerical python have dropped support for oldnumeric which was a module that was made for backward compatibility years ago. Instead open-babel can be used to convert from .pdf format into .pdbqt format. The format is similar to PDB format, but also contains information about what torsions should be active and the assigned charge for each atom. Vina and Smina does not use the same scoring function as Autodock and do not use the charges at all for the default scoring function. Open-babel fast gives us the two needed .pdbqt files.
obabel 1OYT-receptor.pdb -xr -O 1OYT-receptor.pdbqt obabel 1OYT-FSN.pdb -O 1OYT-FSN.pdbqt
Docking with Smina is done from the command line and is very easy to script thanks to the possibility to calculate the box from an existing ligand. The –autobox_ligand and –autobox_add switches are used to define a docking box that is 8Å greater than the ligand specified. The –exhaustiveness 16 switch tells Smina to use more time on finding the best scoring binding mode of the ligand in the binding site, default is 8.
smina.static -r 1OYT-receptor.pdbqt -l 1OYT-FSN.pdbqt --autobox_ligand 1OYT-FSN.pdbqt --autobox_add 8 --exhaustiveness 16 -o 1OYT-redock.pdbqt
After a couple of minutes Smina has docked the ligand. The .pdbqt file can be loaded in Pymol and compared with the already known binding mode.
pymol 1OYT-receptor.pdb 1OYT-redock.pdbqt 1OYT-FSN.pdb
 As can be seen on the image to the right, Smina did a good job of finding the correct binding mode as the top scoring pose. However, the test was a redocking which is the easiest among tests of docking performance. Using other proteins, ligands and PDB file alternatives can give other results. The docking algorithm also has a inherent dependence on the initial states and thus on the random seed, so proceed with caution 😉
As can be seen on the image to the right, Smina did a good job of finding the correct binding mode as the top scoring pose. However, the test was a redocking which is the easiest among tests of docking performance. Using other proteins, ligands and PDB file alternatives can give other results. The docking algorithm also has a inherent dependence on the initial states and thus on the random seed, so proceed with caution 😉
Pingback: Never use re-docking for estimation of docking accuracy | Wildcard Pharmaceutical Consulting
Pingback: Machine Learning optimization of Smina cross docking accuracy | Wildcard Pharmaceutical Consulting
Pingback: Docking with rDock 1OYT | Wildcard Pharmaceutical Consulting
Thank you for your useful post!
Hello, I’m graduated student.
I’m studying docking with autodock tools and chimera in Linux.
I used to seperate ligand and receptor with chimera, and I make the pdbqt files and check grid box in autodock tools and then docking with python(openbabel, rdkit). This process takes lots of times, so I want to use smina.
I’m wondering how to use smina.
I read your code carefully, but I don’t know how to use your code(especially pymol code).
What format, and how to implement that code?
I hope your comment.
Thank you.
OK, There is actually no python code in the blog post. The first code block are Pymol commands, the ones typed directly in the interface, and then the rest of the codeblocks are from the command line (bash).
However, iyt’s also possible to control pymol from a python script/session, but the syntax is a bit differently
Typing “fetch 1OYT” in the command prompt in pymol will fecth the PDB file with ID 1OYT
but in a python session its “pymol.cmd.fetch(‘1OYT’)” after importing and launching the pymol object.
I suggest you read more about it at the PymolWIKI https://pymolwiki.org/index.php/Python_Integration
Thank you for sharing your knowledge. Your post is very helpful!
I’m a newbie in docking. I know that PDB often lack hydrogen atoms. So we need to add polar hydrogen as they are required for correct docking. Could you please clarify why do you add H only to O or N (“h_add elem O or elem N”)? What about C and S? Is it because of non polar bounds between C,S and H?
CH is not considered a polar bond or hydrogen bond donor under normal circumstances. SH hydrogen bonds are also a lot weaker than the ones involving NH and especially OH, so they are also often not considered as such in docking functions like Sminas. If your docking site or ligands contains SH, you can add them with “h_add elem O or elem N or elem S” and check how the performance difference is in your case.
Thank you! It is clear now)
Many Thanks for sharing your knowledge. Your post have very helpful content.
Considering add H to PDB structure, how to avoid misadding H atoms. I use propka but for very large molecules, how to ensure?
Yes, it’s a primitive and error-prone way to add hydrogen atoms to the ligand as well as receptor. Propka may be better finding the right protonation states. I’m not aware of an open-source implementation that is completely error free, given the all the interesting PDBs out there. For proteins I sometimes used Salilabs modeller to remodel the protein.
你好,非常感谢你的分享.
我现在在用smina 的minimize做蛋白配体的能量最小化,当我的蛋白和配体原子形成共价键或者配位键时,应该怎么加限制呢?我没有找到相应的条件,请问你知道么???
感谢您的评论。 我正在尝试回答我的最佳中文,但是不幸的是,我不知道如何在Smina中增加对共价键或复合物的限制。 也许您可以问de Koes?
Many Thanks for sharing your knowledge. Your post have very helpful content.I completed the docking of protein and ligand according to your instructions, and generated a mol format file of the docking result. I want to ask whether smina can output the information of the docked ligand and protein into different files, such as output a ligand.mol file and a protein.mol file?
Thanks for your question. I don’t think Smina can output mol files of the ligand and protein. You could possible use Pymol for that, as you can read in the ligand pdbqt file and save the pose as a molfile. Also openbabel can be very useful for format conversions.
Como puedo correr docking flexible con smina, estoy usando config.txt. Ese archivo tiene el receptor.pdbq , el archivo de aminoácidos flexibles (flex.pdqt) , ligando.pdbq y las coordenadas.
receptor = 3aicFH.pdbqt
flex = flex.pdbqt
ligand = ACR-ct-manual.pdbqt
center_x = 194.05
center_y = 42.996
center_z = 195.375
size_x = 46
size_y = 50
size_z = 48
out = 3aicA-ACR-ct-smina-flex-out.pdbqt
exhaustiveness = 8
num_modes = 10
log = log-ct-3aicA-ACR-smina-flex.txt
El programa corre pero no se agregan las coordenadas de los residuos flexibles
_______ _______ _________ _ _______
( ____ \( )\__ __/( ( /|( ___ )
| ( \/| () () | ) ( | \ ( || ( ) |
| (_____ | || || | | | | \ | || (___) |
(_____ )| |(_)| | | | | (\ \) || ___ |
) || | | | | | | | \ || ( ) |
/\____) || ) ( |___) (___| ) \ || ) ( |
\_______)|/ \|\_______/|/ )_)|/ \|
smina is based off AutoDock Vina. Please cite appropriately.
Weights Terms
-0.035579 gauss(o=0,_w=0.5,_c=8)
-0.005156 gauss(o=3,_w=2,_c=8)
0.840245 repulsion(o=0,_c=8)
-0.035069 hydrophobic(g=0.5,_b=1.5,_c=8)
-0.587439 non_dir_h_bond(g=-0.7,_b=0,_c=8)
1.923 num_tors_div
Using random seed: 373867140
mode | affinity | dist from best mode
| (kcal/mol) | rmsd l.b.| rmsd u.b.
—–+————+———-+———-
1 -9.4 0.000 0.000
2 -9.2 2.825 10.907
3 -9.1 4.702 9.825
4 -9.1 2.217 9.769
5 -8.9 3.331 4.961
6 -8.8 3.092 8.961
7 -8.7 4.037 9.217
8 -8.6 1.995 9.236
9 -8.6 1.931 9.602
10 -8.4 4.876 9.675
Gracias por comentar. Si entiendo bien, ¿no se agregan las coordenadas de los residuos flexibles? Nunca hice un acoplamiento flexible con Smina, así que desafortunadamente no puedo ayudar con eso.
How to prepare Interaction File after Docking?
Thanks for commenting. I don’t understand your question, can you be a bit more specific?
Sorry that message was not posted fully. What I am getting out of docking as per your docking with Smina is a redocked ligand poses. Now I want to see the different h-bonding, hydrophobic etc. interactions between the ligand and target protein. How can I visualize it? In autodock this is done by writing the ligand-protein complex file, but I did not see any command here to prepare the complex. Please tell me how can I do that.
No, Vina/Smina only outputs the poses. You would have to analyze them afterwards in an external program. As example, PyMOL has a command for finding contacts within certain limits (Show Contacts and Show Bumps), maybe UCSF Chimera also has some nifty tools?
Just to add to these suggestions, PLIP (protein-ligand interaction profiler) is a really easy-to-use webserver that will indicate interactions such as h-bonds, hydrophobic interactions, pi-stacking etc.. and display it in an already-annotated PyMOL session file you can download!
Thanks for the comment and tip 🙂
Great post.
What is the logic behind specifying both `-l` and `–autobox_ligand` as 1OYT-FSN.pdbqt? Would these ever be different files?
Hi Jeremy,
Thanks, glad you found it useful. Yes, the ligand to dock and the ligand defining the autobox would be different if you e.g. have a ligand you designed. You would then want the native ligand to define the box, but the ligand for docking would not be anywhere near the binding site and could thus not be used to define the box. Happy docking
Esben
Thanks for sharing such a great post. I would appreciate it, if you could able to share the post for smina virtual screening personally or through the blog. Thanks in advance. Looking forward to hear from you.
I’m not sure what post you are talking about?
Oh sorry, I guess it was not clear question. BTW I’m taking about the “ligand docking with Smina” post. In that post, you only showed how to dock single protein- single ligand docking. My question is, Is it possible for you to provide multi docking approaches using Smina.
I have not worked with Smina and docking for a couple of years now, so I’ll not write such a post now. I guess you could wrap up the commands in a bash script or such, iterating through your directory of ligands, then extract the scores you want with a regular expression in your scripting language of choice. Also, take a look at https://github.com/MolecularAI/DockStream some of my colleagues work, you can probably run it without reinvent integration, simply to do virtual screening.
There’s a tutorial which you may find interesting https://github.com/MolecularAI/DockStreamCommunity/blob/master/notebooks/demo_AutoDock_Vina.ipynb
Hi,
Thanks for the great post!
The following comment is about Openbabel, but I observed that with obabel, the generated pdbqt file has zero charges for all atoms.
I tried explicitly mentioning the charge method; still, all the atoms have zero charges.
“obabel -ipdb ligand.pdb -opdbqt -O ligand.pdbqt -partialcharges gasteiger”
In your experience, is this common behavior with openbabel?
Best,
Mandar Kulkarni
I vaguely remember I had similar issues with openbabel when I used it to make the pdbqt files. Can’t remember if I got it solved. However, if you are going to use the standard Smina docking and scoring function, you don’t need the charges as they are not used by the scoring function.