Machine Learning optimization of Smina cross docking accuracy
In the two previous blog posts Ligand docking with Smina and Never use re-docking for …, it was demonstrated how easy it is to dock a small ligand using Smina, and how deceptively accurate a docking program can be when using re-docking rather that cross-docking.
It is however possible to re-tune the docking function to a specific purpose using machine learning approaches. This is the subject of my recent publication in Computational Biology and Chemistry: http://dx.doi.org/10.1016/j.compbiolchem.2016.04.005
I am quite enthusiastic about the approach. The fact that an alternative parameter set could be automatically derived from a small training set that matched the performance of the default parameters is not an insignificant accomplishment
– Anonymous Reviewer
In the article it’s demonstrated how the docking function can be re-tuned using a surprisingly small training set. In the approach the docking program in itself is treated as a kind of “black box”. A training set of 11 cross docking receptor pairs is fed to the docking program together with custom weights for the docking and scoring function. After docking, the RMSD of the cross-docked ligand is compared to the native pose and a loss function is calculated. In machine learning, a loss function is a function which calculates the “goodness” of the solution. So if good weights are chosen, ligands will be docked with a low RMSD between the docked and the native pose resulting in a low loss function. The loss function is then iteratively minimized with a particle swarm optimization algorithm. Particle swarm optimization is a global optimization algorithm that mimics the behavior of schools of fish and flocking birds to efficiently reach a good solution in a few hundred steps in this case.
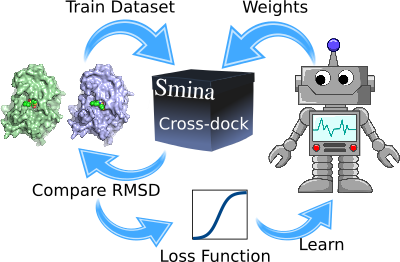
Illustration of Docking Machine Learning algorithm
This resulted in new weights for the Smina program that can be used instead of the default ones. Here’s how to redo the cross-docking experiment from last time using the new weights. First create a new text file with the terms and weights and name it CrossDock.score
-0.0460161 gauss(o=0,_w=0.5,_c=8) -0.000384274 gauss(o=3,_w=2,_c=8) -0.00812176 hydrophobic(g=0.5,_b=1.5,_c=8) -0.431416 non_dir_h_bond(g=-0.7,_b=0,_c=8) 0.366584 repulsion(o=0,_c=8)
Then dock as in the previous blog post, but using the –custom_scoring switch of Smina.
smina.static --custom_scoring CrossDock.score -r 1G32-receptor.pdbqt -l 1OYT-FSN.pdbqt --autobox_ligand 1OYT-FSN.pdbqt --autobox_add 8 --exhaustiveness 16 -o FSN-Crossdock-newWeights.pdbqt pymol FSN-Crossdock-newWeights.pdbqt 1OYT-FSN.pdbqt 1G32-receptor.pdbqt 1OYT-receptor.pdb qt
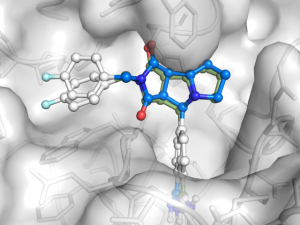
Cross Docking with Optimized Weights
This gives better results than the previous cross-docking although not quite as perfect as the first test using re-docking. The optimized weights are lower for repulsion but higher for the first Gaussian and the hydrogen bond terms. This enables the docking program to dock ligands even in the presence of minor steric clashes. I haven’t tested but the parameters should be compatible with Autodock Vina, although the way the weights are changed are different.
The scoring should not be used to estimate the affinity of the ligand for the receptor. Here the default scoring function is much better than these weights, which are only optimized for cross docking accuracy.
1OYT and 1G32 are a receptor pair which was used in the test set in the publication. Be sure to test whether the default docking function or these weight gives best results for your target of interest. Now you know how to do it 😉 Let me know your experiences.
Best Regards
Esben Jannik Bjerrum
P.S. Drop me a note if you are interested in contributing for the hunt for even better docking functions.
It’s challenging to locate well-informed folks on this topic, but you
sound like you understand what you’re talking about!
Thanks
I’m glad it was useful.
I’d have to check with you here. Which is not something I usually do!
I enjoy reading a post that will get folks think. Also, thanks for enabling me
to comment!
Thank you for commenting.
I am no longer positive the place you’re getting your information, however great topic. I needs to spend a while studying more or figuring out more. Thanks for excellent info I used to be on the lookout for this info for my mission.
Great, let me know if you succeed with your docking studies 😉
Interesting. Very useful blog.
I use smina a lot on daily basis. It is a great and light tool, although not very popular. I have been able to automate and combine with different machine learning scoring functions. the performance so far is great. One thing that I often struggle with is how to optimize smina, or basically any other docking/scoring program for unknown targets, I mean targets there are no co-crystallized ligands exist for. Can we simply do some tricks to accomplish this task based only on the nature of the binding site, for example size, hydrophobicity, charges, etc. my understanding is that the only logical way to do that is using an already crystallized ligand, any ideas are out there?
Thank you for commenting. I have some ideas for further optimization, but nothing I have time to pursue right now. What do you think about starting up a research collaboration? We could discuss and ideate with a Skype meeting?
BR
Esben
Why particle swarm optimization and not a genetic algorithm? Is the later faster than the former? You can also adjust the GA to explore the vicinity of the best solution using crowding…
Hi Thomas,
There is of course a broad range of optimization algorithms available, and I also tested out some different ones. However, PSO was found to be fairly efficient, understood in the sense that it needed a low number of function evaluations to find a good solution. The function evaluation are quite time consuming as there is a need to do several dockings, so efficiency is wanted. I can’t think of a reason why a GA should also not work, It may need more function evaluations, but could also maybe yield a better set of parameters. If you want, we could have a chat over Skype about the possibilities for testing it out. What do you want to use Vina for?