Wildcard scoring function beaten by rDock!

Inspired by the success reported in my last blog post [Link] about the open source docking program rDock [http://rdock.sourceforge.net], I decided to investigate the docking accuracy performance of rDock more in depth.
Once upon a time in the west the commercial docking program GOLD was benchmarked with two test set. The first was a set compiled of 85 receptor-ligand complexes, collected and selected for diversity in the receptor site and ligand characteristics. [http://pubs.acs.org/doi/abs/10.1021/jm061277y]
However, this benchmark was a re-docking exercise, and when it was realized that re-docking gives too optimistic results regarding the perceived docking accuracy a second set was compiled. This was collected by finding the receptors from the first set, which had other entries in the protein data bank co crystallized with a different ligand or being in the APO form. This is the Astex non-native set. [http://pubs.acs.org/doi/abs/10.1021/ci8002254] They are both available for download [https://www.ccdc.cam.ac.uk/support-and-resources/downloads/], and together they forms a an easy way of conducting a cross docking experiment across a range of different receptors.
The test reflect a kind of naive approach, as no post processing and evaluation of the results are done. In reality, a skilled computational chemist will probably get a much higher rate due to clever selection of the docking target, preparation of the docking target by relaxing and minimization, as well as filtering the docking poses by manual inspection. But as a benchmark of what to expect from a docking program, I think it is a more adequate test than just using re-docking.
Thus, I wrote a small python script to interface both Smina and rDock, and collect results from the cross dockings in the form of the RMSD of the best scored pose.
I’m not docking to the entire non native set, but only to the alphanumerically first non-native receptor. This gives 65 docking experiments for rDock, and 59 for Smina, as some of the receptors contained atom types unrecognized by smina.
A few hours later, I could plot the results as this graph.
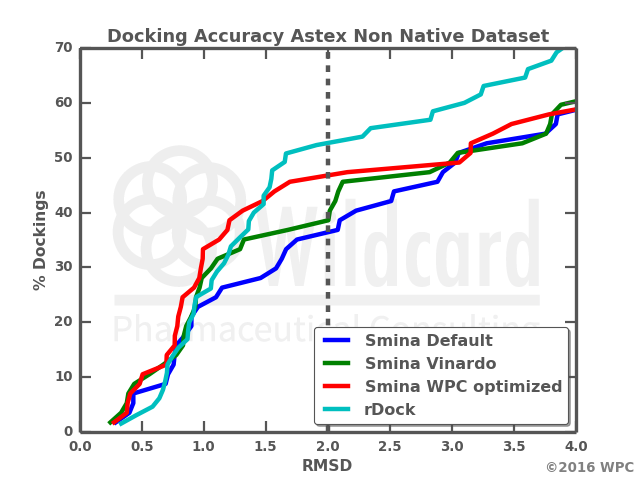
This plot shows the fraction of dockings falling below a given RMSD threshold. This way the ranking can be judged over a range of wanted RMSD accuracies. It is clear that rDock is the best at the usually employed threshold at 2 Ångström, and thus has a larger proportion of the dockings resulting in a best pose closer to the experimentally determined. However, had the threshold been set for 1Å, the rankings would favor the docking function optimized for cross docking studies by Wildcard (WPC) http://www.cheminformania.com/useful-information/machine-learning-optimization-of-smina-cross-docking-accuracy/. At 3 Å both the Vinardo and Vina default scoring function rank better than the by Wildcard optimized docking function. By the way, Vinardo is an alternative docking function for Smina, which by manual fine tuning of the van der Waals radii of the atoms and the terms and parameters, got a much higher success rate in re-docking experiments. Its a pity the authors did not employ a cross docking benchmark in their optimization efforts together with their re docking set. [http://journals.plos.org/plosone/article?id=10.1371%2Fjournal.pone.0155183.]
The simple statistics at 2 Å speak their clear language, and I must see the wildcard optimized score beaten by the default rDock scoring.
- Smina Default
- Percent below 2 A: 0.35
- Smina Vinardo
- Percent below 2 A: 0.37
- Smina WPC optimized
- Percent below 2 A: 0.46
- rDock
- Percent below 2 A: 0.52
Said in other words. A docker using rDock out of the box and naively accepting the best pose as the right one, has a slight bit more chance of being within 2 Å of the experiment, whereas a docker using Smina has a greater risk of being wrong.
I think the reason for this difference is the simplicity of the Vina based scoring function regarding their treatment of hydrogen bonds. Hydrogen bond and polar terms keep coming up in examinations of which term has the greatest discrimination power between correct poses and decoys (WPC internal research).
The hydrogen bond term in vina simply measures the distance between possibly hydrogen bond donors and acceptors, and are thus non-directional. The polar term employed by rDock also takes into account the angle of the interacting groups and are thus probably better at handling and predicting the optimal geometry.
It is however interesting to note the performance of the best placed ligands (<1Å RMSD). Here all Smina based scoring seem to have an edge over rDock, suggesting that they would be better at handling minimization of the docked structures for post-docking work up and analysis.
Reservations
The test is from a single run, and as there is a stochastic element to the dockings the results may change slightly on re testing, so small differences in the benchmark, may turn up the opposite if a different random seed is used.
11 of the ligands from the non-native set were used in the optimization of the WPC score, which may lead to a better performance in the test set. However, it was not the same non-native receptor versions. The same argument could in principle be held against the performance of rDock, as this was tuned against the diverse set. Thus rDock has been trained on all ligands from the non-native set as well. I haven’t cross checked what overlap there is between the non-native set and the sets used for tuning Vina and Vinardo docking functions.
So it is cautiosly that I come with the following rank ordering of the docking programs regarding their docking power:
rDock > WPC > Vinardo > Smina
I have not included the crashed Smina runs in the statistics, but including those would only make the difference between the two different programs greater.
The result is interesting. the Vinardo score was published on Plosone, where it is said the atom radiis had been changed. However, It is better check the source code. On line 7 of builtinscorning.cpp file, the atom radiis are different from those on Plosone. How did you do? which radiis did you used?
Thank you for your interest. I used a version of Smina, where I could see in the version control log, that the radii had been changed by the authors of the Vinardo paper. However, I can give you the exact radii I used in the benchmark, if you want to. What are you going to use Smina/Vina for?
zhong wu da dao 1801 hao
Sorry for a mistake for the reply. I’m curious about the conformation prediction by docking methods. I tried to introduce the vinardo_score’s radii and parameters to vina directly. However, it seems that the results were not satisfied as smina/vinardo.
As I understand it you tried to put the radii in the source code yourself, and got worse results than using the Vinardo scoring switch of Smina? I’ll need a bit more details to understand exactly.
It seems to be popular to tune functions for Vina, heres another study http://www.inderscienceonline.com/doi/pdf/10.1504/IJCBDD.2017.082806