Peeking into the chemical space using free tools
As covered before, chemical space is huge. So it could be nice if this multidimensional molecular space could be reduced and visualized to get an idea about where how query molecules relate to one another. This small tutorial show a simple example of how this could be done with the PCA decomposition from scikit-learn and molecular fingerprints calculated with RDkit.
First the dataset is created from 50000 molecules using RDKit, the usual hashing to a set bit length is skipped, and instead only the bits where theres more than 0.001 of the bits set is kept.
from rdkit import Chem
from rdkit.Chem import AllChem
from rdkit import rdBase, DataStructs
import glob
#Dictionary Based Fingerprints
import sklearn.feature_extraction
files = glob.glob('/home/esben/Zinc_files/*sdf')
fps=[]
def getMorganAsDict(mol):
d = {}
d.update(AllChem.GetMorganFingerprint(mol,2).GetNonzeroElements())
return d
for sdf in files[0:1]:
print "Processing %s"%sdf
sup = Chem.SDMolSupplier(sdf)
for moli in range(0,50000):
try:
mol = sup[moli]
d = getMorganAsDict(mol)
fps.append(d)
except:
print "Error in Conversion"
v = sklearn.feature_extraction.DictVectorizer(sparse=True, dtype=float)
v.fit(fps)
print(len(v.feature_names_))
print(len(v.vocabulary_))
##For modelling
X = v.transform(fps)
#Remove most sparse
from sklearn.feature_selection import VarianceThreshold
cutoff = 0.999
sel = VarianceThreshold(threshold=(cutoff * (1 - cutoff)))
X2 = sel.fit_transform(X)
print X2.shape
#Pickle v, and X.
import pickle
pickle.dump(X2,file('trainingset.pickle','w'))
pickle.dump(v,file('DictVectorizer.pickle','w'))
pickle.dump(sel,file('FeatureSelector.pickle','w'))
This gives a dataset with the most populated bits of the morgan fingerprints.
#Make a PCA and plot:
import pickle
Xo = pickle.load(file('trainingset.pickle','r'))
#Convert to dense array
X = Xo.toarray()
del Xo
print X.shape
#Standardize
from sklearn.preprocessing import StandardScaler
scaler = StandardScaler(copy=False)
scaler.fit_transform(X)
from sklearn.decomposition import RandomizedPCA
pca = RandomizedPCA(n_components=3)
pca.fit(X) #Dense data required
print pca.explained_variance_ratio_
#RandomizedPCA [ 0.00329575 0.00263305 0.00252065]
Xred =pca.transform(X)
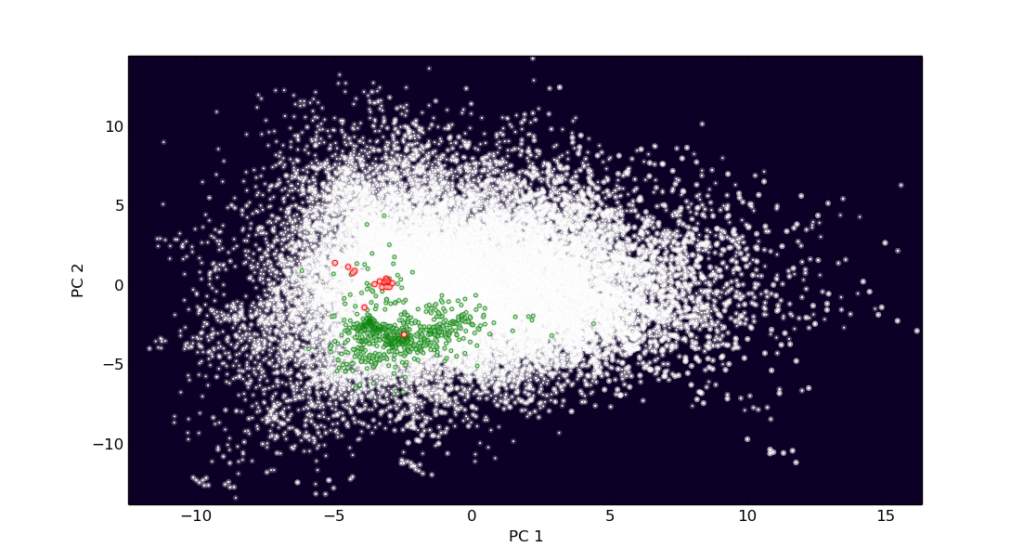
Now the (50000, 4028) matrix has been reduced to a 50000×3 matrix, while trying to preserve most of the variance of the data. Lets try and plot it together with some other datasets which where made to fit.
#Read DHFR test set
Xdhfr = scaler.transform(pickle.load(file('dhfr_testset.pickle','r')).toarray())
ydhfr = pickle.load(file('dhfr_testset_y.pickle','r'))
thr = 1e-2
enc_dhfr0 = pca.transform(Xdhfr[ydhfr < thr])
enc_dhfr1 = pca.transform(Xdhfr[ydhfr > thr])
from matplotlib import pyplot as plt
def star_scatter(X,Y,size,color):
plt.scatter(X, Y, c=color, s=size*2,linewidths=0)
plt.scatter(X, Y, c='w', s=size, linewidths=0)
plt.scatter(X, Y, c=color, s=max((size*4,20)),linewidths=0, alpha=0.2)
star_scatter(Xred[:,0], Xred[:,1],1,'w')
star_scatter(enc_dhfr1[:,0], enc_dhfr1[:,1],5,'g')
star_scatter(enc_dhfr0[:,0], enc_dhfr0[:,1],10,'r')
##Change Color of Canvas
ax = plt.gca()
ax.set_axis_bgcolor((0.05, 0, 0.15))
plt.xlabel('PC 1')
plt.ylabel('PC 2')
plt.show()
I made it look a bit more “space like” on purpose. The green “stars” are the dihydrofolate ligands, with the most active highlighted in red. There seem to be preference for a certain area of the plot, with the high actives preferring a slightly other spot than the bulk of the ligands. However, the explained variance is quite low for the components as the first two components only explain less than 1% of the total variance.
how can we get the file “dhfr_testset_y.pickle”
Hi Dalong,
Thank you for the interest in the Blog post. The DHFR test set is derived from Sutherlands QSAR datasets, http://pubs.acs.org/doi/abs/10.1021/ci034143r, which I had used long time ago. The fingerprints were calculated from the molecules as for the PCA training set, and the ydhfr is a numpy array or vector with the IC50 values. I hope this clarifies the issue, otherwise I can send you the pickle files.
Best Regards
Esben