Learn how to map a simple Ames mutagenicity model to molecular features using RDkit.
 Toxic compounds are most often something that we try to avoid when designing novel pharmaceutical compounds, so it could be nice to get a prediction if a compound is toxic even before ressources are used to synthesize it. But what if it comes back as predicted toxic? Wouldn’t it be great to be able to get feedback about what molecular features of a test molecule that trigger the prediction? It turns out not to be that complicated with RDkit and python, so read on if you are interested in prediction of toxicity. The visualization could also be useful for mapping other features to molecules 😉
Toxic compounds are most often something that we try to avoid when designing novel pharmaceutical compounds, so it could be nice to get a prediction if a compound is toxic even before ressources are used to synthesize it. But what if it comes back as predicted toxic? Wouldn’t it be great to be able to get feedback about what molecular features of a test molecule that trigger the prediction? It turns out not to be that complicated with RDkit and python, so read on if you are interested in prediction of toxicity. The visualization could also be useful for mapping other features to molecules 😉
For the sake of demonstration I found a ~6000 compound data set with associated data from Ames testing. The original research paper is here, where the data is in the supplementary information: DOI: 10.1021/ci900161g
The file contained smiles, CAS number and Smiles and was imported with using the genfromtxt function from Numpy. The comments are set to ‘##’, as smiles triple bonds ‘#’ are otherwise interpreted as starting comments and thus truncating the molecule.
import numpy as np
data = np.genfromtxt("smiles_cas_N6512.smi",delimiter=' \t',names=['Smiles','CAS','Mutagen'], dtype=None, comments='##')
I’ll use the SimilarityMaps module from RDkit for the mapping of model features to the molecules. So I define a small helper script that calculates morgan fingerprints using the Similarity maps module and return them as a numpy array.
from rdkit import Chem, DataStructs from rdkit.Chem.Draw import SimilarityMaps #Helper function to convert to Morgan type fingerprint in Numpy Array def genFP(mol,Dummy=-1): fp = SimilarityMaps.GetMorganFingerprint(mol) fp_vect = np.zeros((1,)) DataStructs.ConvertToNumpyArray(fp, fp_vect) return fp_vect
A for loop structure converts the smiles to RDkit mols and if succesful calculates the fingerprint with the previously defined helper function. 6 smiles failed to convert. For example:
[16:01:18] SMILES Parse Error: syntax error for input: ‘NNC(=O)CNC(=O)\C=N\#N’
The problem is the ‘=N\#N’, RDkit doesn’t think that a double bonded N ‘=N’ can be trans to another N with a triple bond, which I also find a bit strange. It was only 6 smiles that failed to convert so the majority of the data set was converted to fingerprints and are ready for modeling (6506 molecules)
#Convert smiles to RDkit molecules and calculate fingerprints
mols = []
X = []
y = []
for record in data:
try:
mol = Chem.MolFromSmiles(record[0])
if type(mol) != type(None):
fp_vect = genFP(mol)
mols.append([mol, record[1],record[2]])
X.append(fp_vect)
y.append(record[2])
except:
print "Failed for CAS: %s"%record[1]
#See how succesful the conversions were
print "Imported smiles %s"%len(data)
print "Converted smiles %s"%len(mols)
The model built are similar to the one made previously for solubility data. Except here a multiple logistic regression will be used as the prediction is a classification task: Is the molecule mutagenic or not? The regularization parameter C is inversely proportional to alpha as smaller values give more regularization.
from sklearn import linear_model</pre>
<pre>from sklearn import cross_validation</pre>
<pre>#Prepare the data for modelling
X=np.array(X)
y=np.array(y)
y = y== 0
#Cross Validate
X_train, X_test, y_train, y_test = cross_validation.train_test_split(X, y, test_size=0.1)
#prepare scoring lists
fitscores = []
predictscores = []
#prepare a log spaced list of C values to test
Cs = np.logspace(-4, 3, num=10)
#Iterate through Cs and fit
for C in Cs:
estimator = linear_model.LogisticRegression(C = C)
estimator.fit(X_train,y_train)
fitscores.append(estimator.score(X_train,y_train))
predictscores.append(estimator.score(X_test,y_test))
#show a plot
import matplotlib.pyplot as plt
ax = plt.gca()
ax.set_xscale('log')
ax.plot(Cs, fitscores,'g')
ax.plot(Cs, predictscores,'b')
plt.xlabel('C')
plt.ylabel('Mean Accuracy')
plt.savefig('tuneregularization.png')
plt.show()
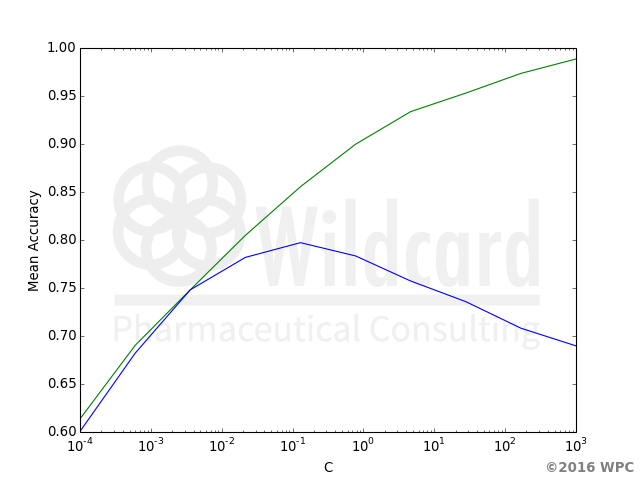
Tuning regularization for a Ames test model using RDkit and SKlearn
The tuning curve shown to the right have an optimum for the test curve (blue) with C around 0.1, 0.05 is chosen for further modeling with the full data set.
#Build a simple model estimator = linear_model.LogisticRegression(C=C) #Fit and score estimator.fit(X,y) print estimator.score(X,y)
The mean accuracy of the model for the fit is 0,82. I’m not sure how this performs in comparison with the one from the original publication as more advanced metrics were used to benchmark the models. However, this fast build and simple model will serve as the base for how to provide feedback from a model to molecular features.
There is no direct way to get from a hashed fingerprint back to the original fingerprints. Hashing functions takes an arbitrarily long bit sequence and convert them to a fixed length. Thus different features can actually in theory set the same bit. And even if we could get the original circular graph elements back, we would still need to assemble the molecule. So we cannot assemble the perfect mutagenic molecule from the feature importance information in the model. Instead the mapping works by calculating a fingerprint for a test molecule and at the same time storing information about what bits are associated with what atoms. Then each atom and the associated bits are removed, and the probability calculated that the molecule is mutagenic. The difference in probability is stored and the same procedure is done for the next atom. This way a weight can be given to each atom if it either adds or subtract to the probability. These weights can then subsequently be mapped to the molecule and hopefully shed light on which parts are the most critical. This functionality is already present and implemented inte SimilarityMaps module in RDkit for some selected fingerprints, including the morgan types.
# helper function
def getProba(fp, predictionFunction):
return predictionFunction(fp)[0][1]
mols = [(Chem.MolFromSmiles('OCC(O)CO'),'Glycerol'),
(Chem.MolFromSmiles('c1ccc2c(c1)cc3ccc4cccc5c4c3c2cc5'),'Benzo[a]pyrene'),
(Chem.MolFromSmiles('CCN(N=O)C(N)=O'),'N-ethyl-N-nitrosourea'),
(Chem.MolFromSmiles('CC(C)CCC(C)CCC1=CC=C2C(=C1)C=CC=C2[N+](=O)[O-]'),'Tester')
]
for mol,name in mols:
#Make Prediction
fp_vect = genFP(mol)
print "Probability %s mutagenic %0.2f"%(name,estimator.predict_proba(fp_vect)[0][0])
#Visualize
weights = SimilarityMaps.GetAtomicWeightsForModel(mol, SimilarityMaps.GetMorganFingerprint, lambda x: getProba(x, estimator.predict_proba))
fig = SimilarityMaps.GetSimilarityMapFromWeights(mol, weights)
fig.savefig(name + '.png', bbox_inches='tight')
The output from the script:
Probability Glycerol mutagenic 0.27
Probability Benzo[a]pyrene mutagenic 0.95
Probability N-ethyl-N-nitrosourea mutagenic 0.89
Probability Tester mutagenic 0.88
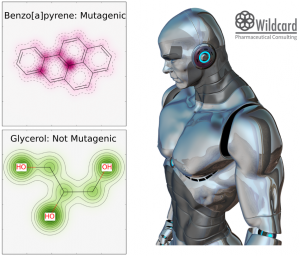
So glycerol doesn’t seem to be mutagenic. Lucky for us. Benzo[a]pyrene is a known mutagen present in coal tar and tobacco smoke. N-ethyl-N-nitrosourea is also a known mutagen and also get a high probability. I didn’t search the data set to see if they are already included and are in fact true predictions or included in the dataset. Mapping the weights from the analysis, color glycerol green and the benzo[a]pyrene red as shown in in the first picture in the blog post. The last one shown to the right is a test molecule I made up. The model predict it to be mostly probable mutagenic.The mapping shown in the picture show that the nitro-naphtalene end is the critical part (colored red), whereas the branched aliphatic chain is not. This fits very well with what I would suspect. The examples are pretty clear, but for other molecules there could be a benefit from getting a bit of feedback from a model. The mapping of the weights gives a glimpse of what is happening inside the model.
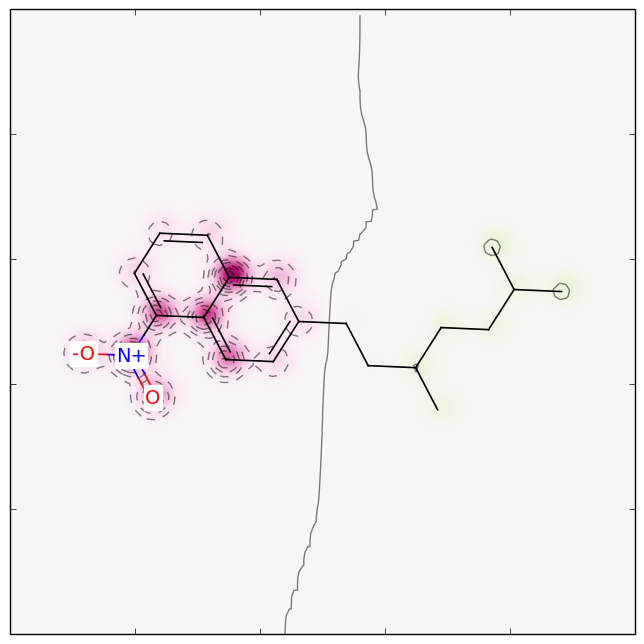
Molecule marked partly mutagenic
For production use I would take more care about the model build. What fingerprints and machine learning models are most relevant? Are there data enough? Are the data normalized and standardized? It would probably also be of value to be able to link back to the most similar compounds in the data set, so the similarities and literature could be evaluated. But then it would take longer than this afternoon. Let me know if it could be useful to your research.
Best Regards
Esben Jannik Bjerrum
FYI: “NNC(=O)CNC(=O)\C=N\#N” isn’t a valid SMILES to start with.
A “/” is a single bond and a “#” is a triple bond. So this says that the two Ns are connected by both a single and a triple bond.
Thank you for your feedback. It wasn’t a critique of RDkit, but these kind of un-valid smiles exists in the wild and in databases and CSV’s, so we need to be able to handle them, by pre-processing or other means. Actually, I only thought the / and \ had to do with cis-trans geometry. That its a single bond I would assume was implicit?